

|
Getting your Trinity Audio player ready...
|
Die Medizinprodukte-Verordnung mit der offiziellen Bezeichnung „EU 2017/745“ stellt Praxis- und gewerbliche Labore vor eine große Herausforderung. Inzwischen steht unmissverständlich fest: Sie tritt am 26. Mai 2020 in Kraft, und zwar für alle Hersteller von Medizinprodukten, und dazu gehören nicht nur gewerbliche Dentallabore, sondern auch Praxislabore. Die Antwort der Bundesregierung auf eine „kleine Anfrage“ der FDP-Bundestagsfraktion im Juni 2019 spricht hierzu unmissverständlich eine klare Sprache: „Ähnlich wie für die Hersteller werden mit der MDR auch die Anforderungen an Sonderanfertiger* erhöht. Besonders hervorzuheben ist hierbei, dass Sonderanfertiger ein Qualitätsmanagementsystem (QMS) gemäß der MDR aufbauen müssen. Besondere Herausforderungen für KMU**, die Sonderanfertigungen herstellen, bestehen in diesem Zusammenhang in den Anforderungen der MDR in Bezug auf die klinische Bewertung (inklusive der klinischen Nachbeobachtung), des Risikomanagements sowie der proaktiven Überwachung nach dem Inverkehrbringen …“
Der Hintergrund
Seit 2008 wurde ein neuer Rechtsrahmen für Medizinprodukte in der Europäischen Kommission diskutiert – 2012 wurden die ersten Entwürfe für den neuen Rechtsrahmen veröffentlicht. Die Grenzen der bis dahin bestehenden Richtlinien (92/43 EWG und deren nationale Medizinproduktegesetze) offenbarten sich in erster Linie durch diverse Probleme mit Implantaten wie beispielsweise Metall- Metall-Hüftprothesen. Mechanisch-korrosiver Abrieb in diesen Prothesen führte zu erhöhten Konzentrationen von Kobalt- und- Chrom-Ionen im Gewebe. Der Hersteller DePuy hatte aus diesem Grund 2010 betroffene Produkte zurückgerufen. Vorangetrieben wurde die Änderung des Rechtsrahmens in erster Linie jedoch durch die Offenlegung einer der größten bisher dagewesenen Medizinprodukteskandale mit etwa 400.000 betroffenen Personen in 65 Ländern – dem Brustimplantat-„PIP-Skandal“ rund um das französische Unternehmen Poly Implant Prothèse, kurz PIP, mit gerissenen oder undichten Implantaten.
Als Reaktion der Europäischen Kommission auf diesen Skandal wurde die 2008 angekündigte Revision des Rechtsrahmens für Medizinprodukte beschleunigt. Die ersten Entwürfe für Verordnungen für Medizinprodukte und In-vitro-Diagnostika wurden am 26. September 2012 von der Europäischen Kommission präsentiert, mit dem vorläufigen Ziel der Übernahme des Entwurfes als Gesetzestext bis Ende 2014. Bis zur endgültigen Verabschiedung der neuen Verordnung dauerte es jedoch bis April 2017.
Es handelt sich dabei um eine Verordnung, die den Schutz des Verbrauchers bzw. des Patienten in ihren Fokus stellt. Es werden europaweit alle medizinischen Wirtschaftsakteure und Gesundheitseinrichtungen miteinbezogen, die für den gesamten Lebenszyklus eines Medizinprodukts verantwortlich sind. Das heißt konkret: Jeder, der am Wertschöpfungsprozess eines Medizinproduktes beteiligt ist, ist für die Vor- und die nachfolgende Stufe (mit-)verantwortlich.
Hersteller von Zahnersatz
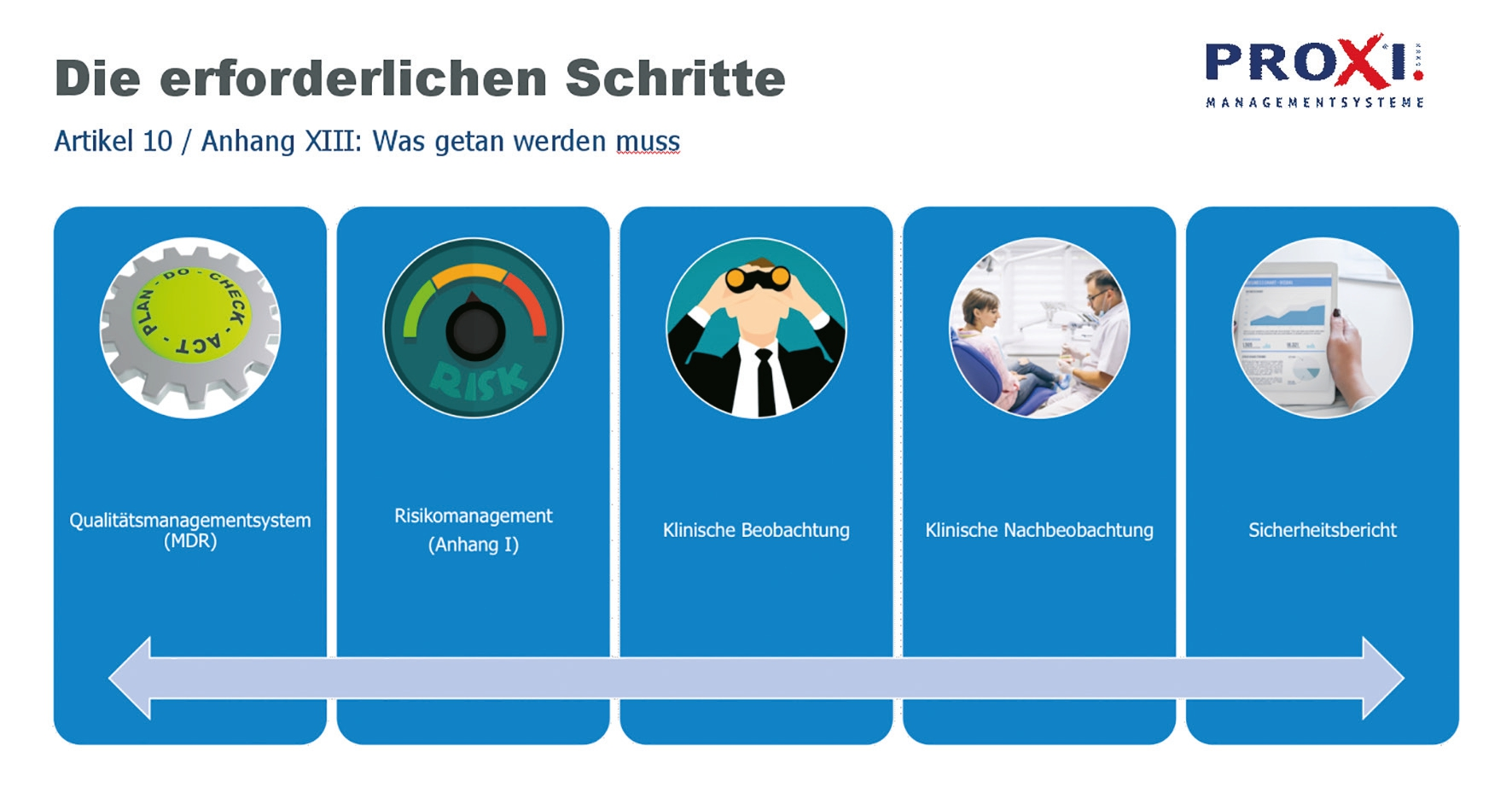
Die MDR unterscheidet nicht zwischen Herstellern von Serienprodukten einerseits und Sonderanfertigungen andererseits. Zunächst existieren nur „Hersteller“. Für Sonderanfertigungen z. B. von Zahntechnikern gelten bestimmte Besonderheiten und Einschränkungen, gegenüber Serienprodukten bleibt jedoch die vereinfachte Konformitätsbewertung bestehen. Das Verfahren für Sonderanfertigungen wird in Anhang XIII MDR beschrieben. Somit sind vor allem der Artikel 10 (Hersteller, Abb. 1) und Anhang XIII (Sonderanfertiger) von zentraler Bedeutung für gewerbliche und Praxislabore.
 PROXI.GMBH
PROXI.GMBHDie Dokumentationspflichten
Die Dokumentationspflichten für Sonderanfertigungen ergeben sich aus Anhang XIII Abschnitt 1 MDR. Es muss wie bisher eine Erklärung erstellt werden, die folgende Angaben enthält:
- Name und Anschrift des Herstellers sowie aller Fertigungsstätten;
- eine Erklärung, dass das Produkt ausschließlich für einen bestimmten Patienten oder Anwender bestimmt ist, der durch seinen Namen, ein Akronym oder einen numerischen Code identifiziert wird;
- Name des Verordnenden und gegebenenfalls Name der betreffenden medizinischen Einrichtung;
- die spezifischen Merkmale des Produkts, wie sie in der Verordnung angegeben sind;
- eine Erklärung, dass das betreffende Produkt den grundlegenden Sicherheits- und Leistungsanforderungen gemäß Anhang I MDR entspricht, und gegebenenfalls ein Verweis auf die grundlegenden Sicherheits- und Leistungsanforderungen, die nicht vollständig eingehalten wurden, mit Angabe der Gründe – das bedeutet deutlich weitergehende Angaben als bisher;
- Die Erklärung muss für einen Zeitraum von mindestens zehn Jahren nach dem Inverkehrbringen des Produkts aufbewahrt werden.
So weit nicht viel Neues. Neben der Erklärung muss der Hersteller von Sonderanfertigungen eine Dokumentation über die Sonderanfertigung erstellen und für die zuständigen Behörden bereithalten. In dieser müssen neben der Fertigungsstätte bzw. den Fertigungsstätten auch Angaben erfolgen, aus denen die Auslegung, die Herstellung und die vorgesehene Leistung des Produkts hervorgehen, sodass sich beurteilen lässt, ob es den Anforderungen der MDR entspricht.
Empfehlung:
- Führen Sie eine sorgfältige Dokumentation der Sonderanfertigung. Archivieren Sie Auftragszettel, Dokumente der Vorprodukte und Materialien usw. – einschließlich der Angaben aus der Abstimmung mit dem Zahnarzt und dem Patienten – sorgfältig.
- Erfassen Sie alle im Produkt verwendeten Materialien, Vorprodukte und Passteile, sodass diese für den jeweiligen Patientenfall nachvollziehbar sind.
- Führen Sie ein System zur Chargenrückverfolgung (z. B. über Lieferscheine, Chargen- und Losnummern).
- Nutzen Sie ggf. vorhandene Aufkleber für Passteile für Ihre interne Dokumentation.
- Bewahren Sie Lieferscheine mit Angaben zu Materialien, Chargen- und Losnummern auf.
Die neuen Anforderungen an Hersteller: Artikel 10 MDR
Was die zukünftig gesetzlich vorgeschriebenen Anforderungen konkret beinhalten, zeigt Abbildung 2 in der Übersicht.
 PROXI.GMBH
PROXI.GMBH1. Qualitätsmanagementsystem:
Das Qualitätsmanagementsystem (QM) ist ein Konzept zur Einhaltung der Regulierungsvorschriften, das die Einhaltung der Konformitätsbewertungsverfahren und der Verfahren für das Management von Änderungen an den vom System erfassten Produkten mit einschließt. Es muss enthalten:
- Verantwortlichkeit der Leitung
- Ressourcenmanagement, einschließlich der Auswahl und Kontrolle von Zulieferern und Unterauftragnehmern
- Risikomanagement gemäß Anhang I
- eine klinische Bewertung
- klinische Nachbeobachtung nach dem Inverkehrbringen
- Produktrealisierung einschließlich Planung, Auslegung, Entwicklung, Herstellung und Bereitstellung von Dienstleistungen
- Implementierung und Management eines Systems zur Überwachung nach dem Inverkehrbringen gemäß Artikel 83 und Prüfung und Dokumentation der Erfahrungen nach dem Inverkehrbringen laut Anhang XIII MDR; usw.
Die MDR schreibt keine spezielle Norm und auch keine Zertifizierungsverpflichtung vor. Jedes gewerbliche und Praxislabor ist jedoch verpflichtet, ein dynamisches und vor allem „gelebtes“ QM einzuführen, aufrechtzuerhalten und ständig zu verbessern (siehe hierzu Artikel 10 Abs. 9).
Empfehlung:
Sofern Sie bereits über ein (zertifiziertes) QM-System verfügen, beachten Sie bitte, dass dieses nicht automatisch alle Anforderungen der MDR erfüllt. Überprüfen Sie Ihr QM-System insbesondere auf die Anforderungen der MDR zur klinischen Bewertung und zum Risikomanagement.
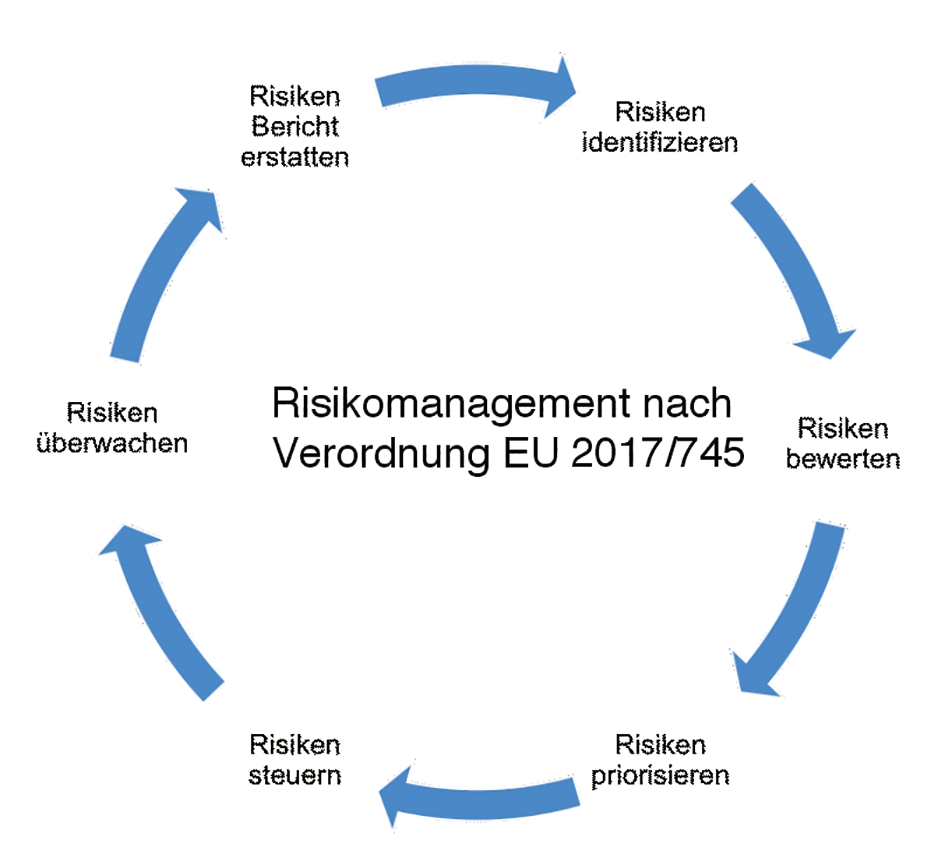
2. Risikomanagementsystem (Anhang I MDR):
Die MDR fordert ein Risikomanagementsystem als kontinuierlichen iterativen Prozess während des gesamten Lebenszyklus eines Produkts, der eine regelmäßige systematische Aktualisierung erfordert. Folgende Aufgaben sind dabei zu erfüllen:
- Einen Risikomanagement-Plan für jedes Produkt (bzw. „Produktfamilie“) festlegen und dokumentieren;
- Risiken identifizieren, analysieren, bewerten, beseitigen und kontrollieren;
- die Auswirkungen der in der Fertigungsphase und durch das System zur Überwachung nach dem Inverkehrbringen gewonnenen Informationen sind bezüglich folgender Aspekte zu bewerten:
– Gefährdungen und deren Häufigkeit,
– Abschätzung der verbundenen Risiken sowie des Gesamtrisikos,
– das Nutzen-Risiko-Verhältnis, – die Risikoakzeptanz, und gemäß den neuen Anforderungen anzupassen.
Hinweis:
Hersteller müssen im Rahmen der MDR die Konformität aller Produktbestandteile nachweisen. Hersteller von Medizinprodukten haben künftig zu gewährleisten, dass ihre Produkte bei Inverkehrbringen den Anforderungen der MDR genügen. Falls sie das nicht können, besteht ein Verkehrsverbot.
3. Klinische Bewertung:
Auch Sonderanfertigungen müssen einer klinischen Bewertung unterzogen werden und sind Teil des QM-Systems. Der Umfang des klinischen Nachweises muss den Merkmalen des Produkts und seiner Zweckbestimmung angemessen sein. Hierzu sind die folgenden Punkte zu berücksichtigen:
- Kritische Bewertung der einschlägigen derzeit verfügbaren wissenschaftlichen Fachliteratur über Sicherheit, Leistung, Auslegungsmerkmale und Zweckbestimmung des Produkts,
- eine kritische Bewertung der Ergebnisse aller verfügbaren klinischen Prüfungen,
- eine Berücksichtigung der gegebenenfalls derzeit verfügbaren anderen Versorgungsoptionen für diesen Zweck.
4. Klinische Nachbeobachtung:
Für die klinische Nachbeobachtung nach dem Inverkehrbringen muss ein Plan festgelegt werden. Darin müssen u. a. folgende Methoden und Verfahren für das Sammeln und Bewerten der Daten beschrieben werden:
- Ermittlung zuvor unbekannter Nebenwirkungen (z. B. Veränderungen infolge von Wechselwirkungen mit Medikamenten)
- Bewertung der klinischen Daten zu gleichartigen oder ähnlichen Produkten | Einholung des Feedbacks von Anwendern
- Durchsicht wissenschaftlicher Fachliteratur und anderer Quellen klinischer Daten.
Der Hersteller hat aus den Erfahrungen der klinischen Nachbeobachtung angemessene Vorkehrungen zu treffen, um erforderliche Korrekturen durchzuführen. In diesem Zusammenhang muss nach Artikel 87 Absatz 1 den zuständigen Behörden jedes schwerwiegende Vorkommnis oder jede Sicherheitskorrekturmaßnahme im Feld oder beides gemeldet werden, sobald der Hersteller davon erfährt.
Die Ergebnisse aus der klinischen Nachbeobachtung nach dem Inverkehrbringen sind in einem Bewertungsbericht über die klinische Nachbeobachtung nach dem Inverkehrbringen zu dokumentieren. Es sind angemessene Vorkehrungen zu treffen, um erforderliche Korrekturen durchführen zu können.
5. Sicherheitsbericht:
Jeder Hersteller von Zahnersatz muss für jede Produktkategorie oder Produktgruppe einen regelmäßig aktualisierten Bericht über die Sicherheit („Sicherheitsbericht“) erstellen. Die Hersteller von Produkten der Klasse IIa (also von Zahnersatz) aktualisieren den Sicherheitsbericht bei Bedarf, mindestens jedoch alle zwei Jahre.
Medizinprodukte-Anpassungsgesetz-EU (Referentenentwurf)
Das Bundeskabinett hat am 06. November 2019 den Entwurf eines Gesetzes zur Anpassung des nationalen Medizinprodukterechts (Medizinprodukte-EU-Anpassungsgesetz – MPEUAnpG) an die Medizinprodukteverordnung 2017/745 beschlossen. Der Gesetzentwurf dient in erster Linie der technischen Anpassung des nationalen Medizinprodukterechts an die neuen EU-Vorgaben.
Das im Referentenentwurf vorliegende neue Gesetz hat hinsichtlich der Anforderungen an die Hersteller von Sonderanfertigungen keine Erleichterungen zum Inhalt. Dargelegt werden in diesem Gesetzentwurf zum Beispiel die Verantwortlichkeiten der Medizinprodukteberater. Auch werden Strafrahmen, Höhe der Strafen und Bußgelder für Verstöße spezifiziert. So ist z. B. das Inverkehrbringen abgelaufener Materialien oder eine nicht erfolgte Meldung eines meldepflichtigen Vorkommnisses durch den Medizinprodukteberater in Zukunft mit bis zu 30.000 EUR Bußgeld bewertet.
Ein Bußgeld droht auch, wenn keine oder eine nicht vollständige Konformitätserklärung nach Anhang XIII (MDR) beifügt wird. Eine Konformitätserklärung ist bereits und wird in Zukunft noch deutlicher zu einem Rechtsakt, der nicht zu unterschätzen ist. Das Labor muss darin bestätigen, dass es den Anhang I (MDR) erfüllt und somit ein Risikomanagement als kontinuierlichen iterativen Prozess während des gesamten Lebenszyklus eines Produkts (Produktfamilie) mit regelmäßiger systematischer Aktualisierung in seinem Labor nicht nur eingeführt hat, sondern auch anwendet.
In weiteren Artikeln wird der Autor die einzelnen gesetzlichen Anforderungen noch weiter vertiefen.
*Zahntechniker gelten als „Sonderanfertiger“
**kleine und mittelständische Unternehmen
Alles Wissenswerte zur neuen EU-Medizinprodukte-Verordnung finden Sie auch ausführlich und in kompakter Form auf www.mdr2020.de
Teil 2
Die Medical Device Regulation (MDR), die neue europäische Medizinprodukte-Verordnung, tritt Ende Mai dieses Jahres in Kraft. Was auf Zahntechniker dabei zukommt, hat unser Autor Karl-Heinz Martiné in Teil 1 dieser Beitragsreihe zusammengefasst. Im Folgenden vertieft er die Anforderungen an das Qualitätsmanagement nach den Vorgaben der MDR.
Bereits nach der derzeitigen noch gültigen Richtlinie 93/42 EWG unterliegen Medizinprodukte strengen Anforderungen an die Qualitätssicherung. Wenn auch bisher eine Verpflichtung zur Qualitätssicherung (nicht zu verwechseln mit Qualitätsmanagement!) bestand, so war bis dato nicht eindeutig festgelegt, welche Inhalte ein solches Qualitätssicherungssystem aufzuweisen hat. Somit wurde oftmals der Nachweis zur Erfüllung dieser Forderung mit der Bescheinigung der Meisterprüfung im Zahntechniker- Handwerk, mit internen Laufzetteln, auf denen die bearbeiteten Positionen standen, und einer dokumentierten Endkontrolle durch eine dazu befugte Person erbracht. So weit der Status quo.
„Gelebtes“ Qualitätsmanagement wird Pflicht
 Martiné
MartinéDies hat sich in der Verordnung EU 2017/745 (MDR) grundlegend geändert, und zwar unabhängig von der Größe eines Unternehmens und der Menge der hergestellten Sonderanfertigungen. Ab 26. Mai besteht für alle Hersteller die Pflicht, ein vollständiges Qualitätsmanagementsystem einzurichten, zu dokumentieren, anzuwenden, aufrechtzuerhalten, ständig zu aktualisieren und kontinuierlich zu verbessern (siehe hierzu Artikel 10 Verordnung EU 2017/745).
Hersteller für Sonderanfertigungen sind in unserer Branche alle gewerblichen Dentallabore, alle Praxislabore, die Medizinprodukte, die im Mund verbleiben, herstellen, und Zahnärzte, die Chairside- Leistungen erbringen (da es sich auch hierbei um die Fertigung von Medizinprodukten handelt).
Auch ist es mit einer einmaligen Dokumentation oder einem Ordner, der erstellt wird und dann im Regal verstaubt, in Zukunft nicht mehr getan. Die Nachweispflicht fordert ein „gelebtes“ Qualitätsmanagement.
Was muss das QM-System beinhalten?
Der wesentliche Unterschied zwischen einem Qualitätssicherungssystem (QS-System) und einem Qualitätsmanagementsystem (QM-System) besteht darin, dass sich ein QS-System auf die Sicherung der Produktion und die jeweiligen Produkte bezieht. Ein QM-System bezieht das gesamte Unternehmen und alle Prozesse der Leistungserstellung mit ein. Hierzu fordert die neue Verordnung in Artikel 10: „Das Qualitätsmanagementsystem umfasst alle Teile und Elemente der Organisation eines Herstellers, die mit der Qualität der Prozesse, Verfahren und Produkte befasst sind. Es steuert die erforderliche Struktur und die erforderlichen Verantwortlichkeiten, Verfahren, Prozesse und Managementressourcen zur Umsetzung der Grundsätze und Maßnahmen, die notwendig sind, um die Einhaltung der Bestimmungen dieser Verordnung zu erreichen.“
Im Gegensatz zu der bisherigen Richtlinie konkretisiert die MDR auch die Inhalte, die ein gesetzeskonformes QM-System mindestens aufzuweisen hat. Hierzu zählt die MDR für Sonderanfertiger insgesamt 11 relevante Punkte auf:
1. Der Hersteller muss entsprechend den Anforderungen der MDR ein dokumentiertes QM-System vorhalten, in dem die zu dokumentierenden Verfahren, Tätigkeiten und Regelungen beschrieben sind, welche die Einhaltung der MDR zum Ziel haben.
– Die Forderung der MDR: „[…] ein Konzept zur Einhaltung der Regulierungsvorschriften, was die Einhaltung der Konformitätsbewertungsverfahren und der Verfahren für das Management von Änderungen an den von dem System erfassten Produkten miteinschließt;“
2. Der Hersteller muss definieren, welche Inhalte des Anhang I auf seine Produkte zutreffen und einzuhalten sind. Dies ist für ein Dentallabor in Anhang I mindestens Abschnitt 3 und Abschnitt 4.
– Die Forderung der MDR: „[…] die Feststellung der anwendbaren grundlegenden Sicherheits- und Leistungsanforderungen und die Ermittlung von Möglichkeiten zur Einhaltung dieser Anforderungen;“
3. Die Geschäftsleitung muss ihre Verpflichtung bezüglich der Entwicklung und Implementierung des Qualitätsmanagementsystems und der Aufrechterhaltung von dessen Wirksamkeit nachweisen, indem sie:
a) ihren Mitarbeitern die Bedeutung der Erfüllung der Kundenanforderungen sowie der anwendbaren regulatorischen Anforderungen vermittelt;
b) eine Qualitätspolitik festlegt;
c) sicherstellt, dass Qualitätsziele festgelegt werden;
e) sicherstellt, dass eine für die Einhaltung der regulatorischen Vorgaben „verantwortliche Person“ im Unternehmen verfügbar ist.
– Die Forderung der MDR: „[…] die Verantwortlichkeit der Leitung;“
4. Ein wesentliches Prinzip der MDR ist das umfassende Ressourcenmanagement hinsichtlich Personal, Maschinen, Materialien und Unterauftragnehmern. Jeder Akteur im Wertschöpfungsprozess eines Medizinproduktes ist für die Stufe davor und die Stufe danach (mit-)verantwortlich. Das bedeutet, er muss kontrollieren, ob sein Lieferant die Bestimmungen der MDR einhält (hierzu ist z. B. der Nachweis einer Zertifizierung nach DIN EN ISO 13485 eine geeignete Methode). Auch muss er im Rahmen seines Risikomanagements bereits bei der Fertigung die Anwendung in der nächsten Stufe im Blick behalten.
– Die Forderung der MDR: „[…] das Ressourcenmanagement, einschließlich der Auswahl und Kontrolle von Zulieferern und Unterauftragnehmern;“
5. Ein elementarer Punkt im Rahmen eines Qualitätsmanagements ist das Risikomanagement nach Anhang I MDR. Immerhin wird mit jeder Konformitätsbestätigung durch den Hersteller (gewerbliches Labor, Praxislabor und chairside) die Einhaltung dieses Anhangs rechtsverbindlich dem Patienten garantiert. Hierbei ist vor allem darauf zu achten, dass dieses Risikomanagement nicht statisch sein darf! Mit einer einmal erstellten Liste ist es ausdrücklich nicht getan. Hierzu Abschnitt 3 Anhang I: „Das Risikomanagement ist als kontinuierlicher iterativer Prozess während des gesamten Lebenszyklus eines Produkts [Produktart] zu verstehen, der eine regelmäßige systematische Aktualisierung erfordert.“
– Die Forderung der MDR: „[…] das Risikomanagement gemäß Anhang I Abschnitt 3;“
6. Schwierigkeiten dürfte vielen Herstellern von Sonderanfertigungen die Forderung nach einer „klinischen Bewertung“ und der „klinischen Nachbeobachtung“ bereiten. Die „klinische Bewertung“ ist die Analyse und Bewertung von klinischen Daten zu einem bestimmten Medizinprodukt mit dem Ziel, dessen Leistung und Sicherheit in der klinischen Anwendung nachzuweisen. Sie erfolgt in der Regel für Sonderanfertigungen anhand von klinischen Daten aus der wissenschaftlichen Literatur. Bei der „klinischen Nachbeobachtung“ besteht nicht nur die Verpflichtung der Befragung seiner Kunden, sondern auch zur „aktiven Marktüberwachung“ durch eine regelmäßige Recherche in einschlägigen Datenbanken wie z. B. beim BfArM (Bundesinstitut für Arzneimittel und Medizinprodukte).
– Die Forderung der MDR: „[…] die klinische Bewertung gemäß Artikel 61 und Anhang XIV einschließlich der klinischen Nachbeobachtung nach dem Inverkehrbringen;“
7. Die Produktion und die Dienstleistungserbringung müssen so geplant, durchgeführt, überwacht und gelenkt werden, dass sichergestellt ist, dass das jeweilige Produkt nur noch ein äußerst niedriges Risiko aufweist. Hierzu müssen z. B. die folgenden Verfahren geregelt werden:
a) Gesicherte Einhaltung der Vorgaben aus den Gebrauchsanweisungen;
b) Kalibrierungsüberwachung qualitätsrelevanter Maschinen und Geräte;
c) Lückenlose Rückverfolgbarkeit der eingesetzten Materialien (Chargenverwaltung), hierzu müssen z. B. bei Rückrufen von verarbeiteten Materialien der jeweilige Zahnarzt und sein Patient informiert werden können.
– Die Forderung der MDR: „[…] die Produktrealisierung einschließlich Planung, Auslegung, Entwicklung, Herstellung und Bereitstellung von Dienstleistungen;“
8. Der Hersteller muss im Rahmen seines QM-Systems regelmäßig seine Kunden hinsichtlich Auffälligkeiten seiner gefertigten Sonderanfertigungen befragen, Reklamationen qualitativ auswerten, die Ergebnisse dokumentieren und bei erforderlichen Präventiv- oder Korrekturmaßnahmen geeignete Maßnahmen ergreifen.
– Die Forderung der MDR: „[…] die Aufstellung, Anwendung und Aufrechterhaltung eines Systems zur Überwachung nach dem Inverkehrbringen gemäß Artikel 83;“
9. Der Hersteller muss Regelungen dafür treffen, dass er durch eine aktive Kommunikation bzw. sein Informationsverhalten Kenntnisse über sicherheitsrelevante Fakten seiner Medizinprodukte bzw. zu verarbeitenden Materialien erhält.
– Die Forderung der MDR: „[…] die Kommunikation mit den zuständigen Behörden, Benannten Stellen, weiteren Wirtschaftsakteuren, Kunden und/oder anderen interessierten Kreisen;“
10. Der Hersteller ist verpflichtet, ein Verfahren für die Bereitstellung von Meldungen an die entsprechenden Regulierungsbehörden einzurichten und zu dokumentieren. Für die Regulierung der Medizinprodukte sind in der BRD die Bundesländer zuständig.
– Die Forderung der MDR: „[…] die Verfahren für die Meldung von schwerwiegenden Vorkommnissen und Sicherheitskorrekturmaßnahmen im Feld im Rahmen der Vigilanz;“
11. Dies sollte eine Selbstverständlichkeit und somit bereits in jedem Dentallabor implementiert worden sein: ein aktives „Reklamations- und Verbesserungsmanagement“. Hier geht es um eine qualitative Auswertung der Reklamationen und nicht nur um die übliche quantitative Bewertung. Die Ergebnisse der qualitativen Auswertung bilden die Grundlage für die Verbesserungen der Verfahren, Materialien, Qualifikation der Techniker usw.
– Die Forderung der MDR: „[…] das Management korrektiver und präventiver Maßnahmen und die Überprüfung ihrer Wirksamkeit und Verfahren zur Überwachung und Messung der Ergebnisse, Datenanalyse und Produktverbesserung.“
Fazit
Wie den beschriebenen Anforderungen zu entnehmen ist, wird den Herstellern von Sonderanfertigungen nun ein umfassendes Qualitätsmanagementsystem abverlangt. Die DIN EN ISO 13485 gibt zur Erfüllung der Anforderungen aus der MDR eine gute Grundlage und Struktur vor. Es spielt hierbei keine Rolle, ob dieses QM-System von einer externen Stelle zertifiziert wird oder nicht, die MDR schreibt lediglich die in diesem Beitrag beschriebenen Anforderungen vor. Unser Beratungsunternehmen hat in den vergangenen 20 Jahren die Erfahrung gemacht, dass auch das zunächst beste QM-System nicht am Leben gehalten werden kann, wenn es nicht von der Geschäftsleitung mitgetragen und aktiv unterstützt, ständig gepflegt und das Unternehmen von fachkompetenten Beratern gut betreut wird.
Den kompletten Gesetzestext der MDR finden Sie z. B. unter www.eur-lex.europa.eu unter Eingabe von EU 2017/745 im Suchfeld.
Alles Wissenswerte zur neuen EU-Medizinprodukte-Verordnung finden Sie auch ausführlich und in kompakter Form auf www.mdr2020.de






 Mit Google einloggen
Mit Google einloggen
 Mit Facebook einloggen
Mit Facebook einloggen
Keine Kommentare.